Optimizing the transition state geometry in vacuo, starting with a good guess.
We will start by optimizing the transition state of the Diels-Alder reaction in vacuo at the semi-empirical PM3 level. This is easy since we already have the struture of the transition state analogue. We simply take the coordinates of that analogue from the x-ray structure (1C1E.pdb) and modify them a bit, such that we get the appropriate input coordinates for a Gaussian98 transition-state optimization calculation. We will use the semi-empirical PM3 hamiltonian throughout this tutorial, because of its cost-efficiency. Note that in the x-ray structure the -R group of the analogue (Figure 1) is not resolved. We ignore that group for the moment and focus on the system with -R = -CH3.
The following steps will lead us to the transition state geometry of the reaction in vacuo.
- We download 1C1E.pdb
x-ray structure from the pdbdatabase
- We use rasmol to isolate the transition state analogue (ENH)
- open 1C1E.pdb with rasmol
localhost:~>rasmol 1C1E.pdb
- select the TS analogue
RasMol>restrict ENH
- center the analogue
RasMol>center selected
- zoom in on the analogue
RasMol>zoom 600
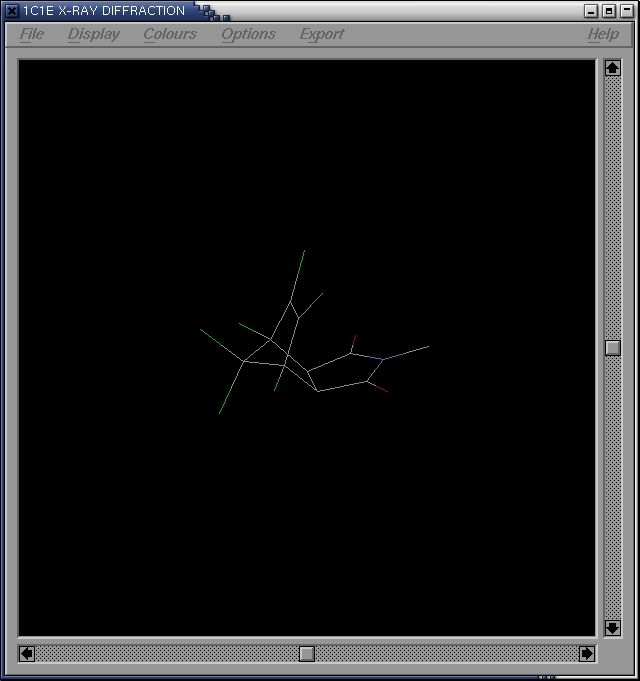
- save the analogue in a seperate pbd-file
Rasmol>save analogue.pdb
- open 1C1E.pdb with rasmol
- We use MOLDEN to create an gaussian inputfile for a
transition state optimization from analogue.pdb
- open analogue.pdb with MOLDEN
localhost:~>molden -P analogue.pdb (with the -P option MOLDEN expects pdb format as input)
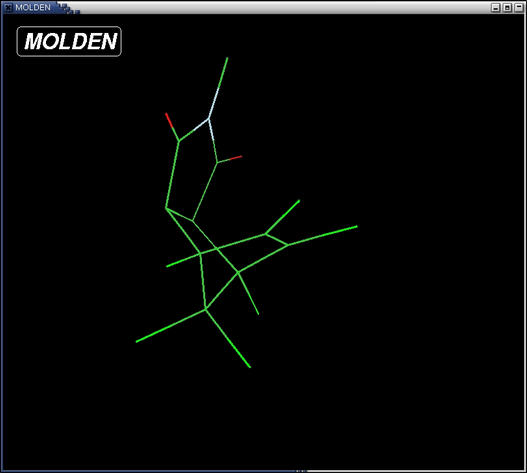
- click the stickcolor button in the Molden Control window to
display the atoms in different colors
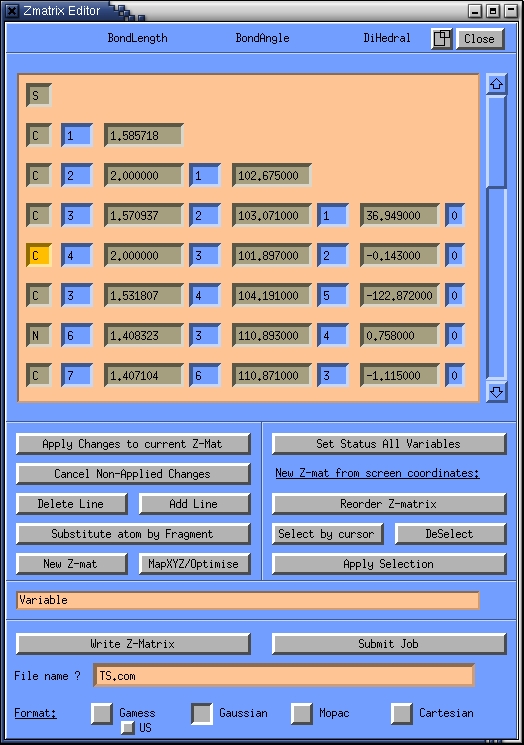
- click on the Zmat editor button to enter the Z-matrix
editor
- click on the "select by cursor" button in the Zmat editor and
select all atoms in the molecule visualiztion window by draging a box
around them with the mouse pointer. Click the "apply selection" button
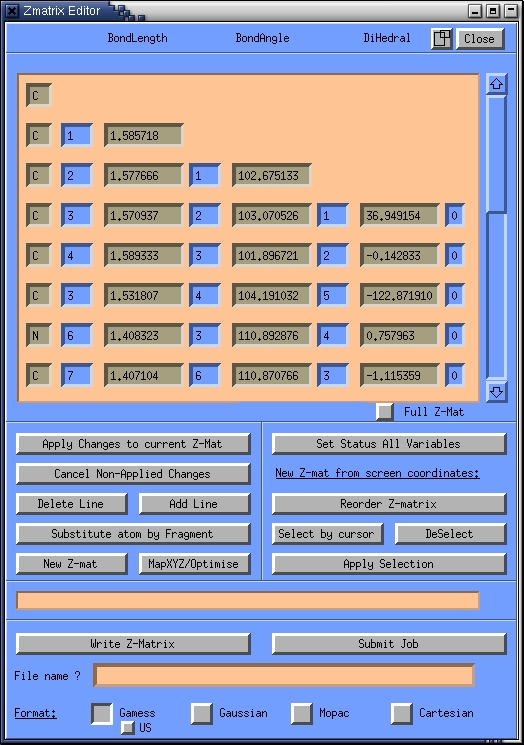
to complete the selection. A Z-matrix represents the geometry in terms
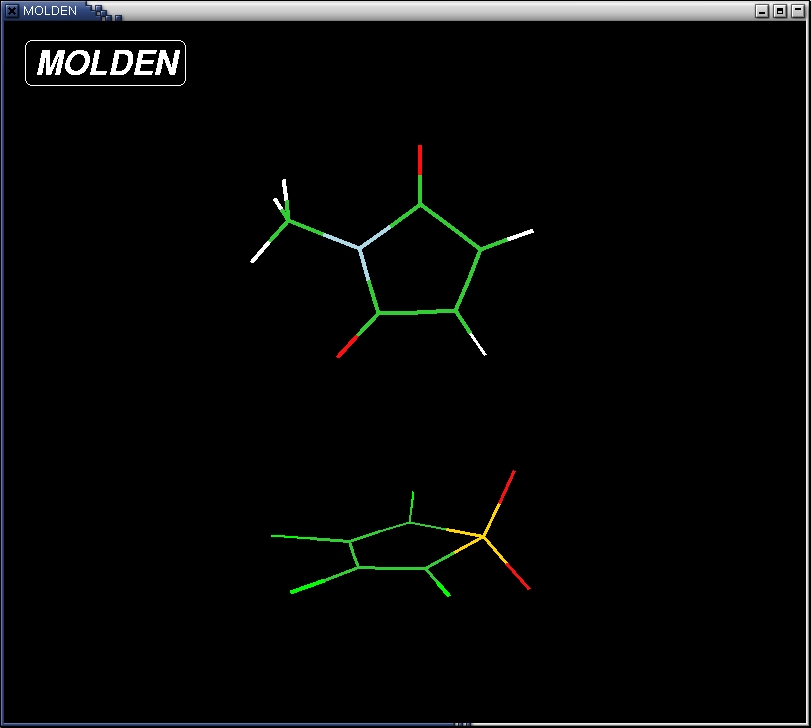
of distances, angles and torsions. For instance, atom 7, is a Nitrogen
atom. The distance of this atom to atom 6 is A; the angle between this
atom and atoms 6 and 3 is 110 degrees and the torsion angle between
this atoms and atoms 6, 3 and 4 is 0.757 degrees.
- increase a bit the lengths of the two bonds that are supposed to
be formed in the Diels-Alder reaction (C4-C5 and C2-C3). Change the
values of the bondlengths of C5-C4 and C3-C2 to, say 2 angstrom in the
Z-matrix. Now the molecule resembes the actual Transition State a bit
better already.
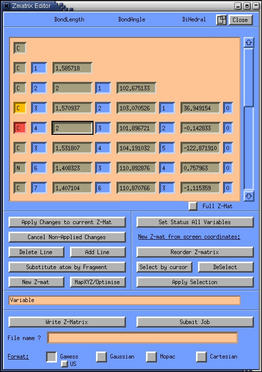
- next, change the atoms of the analogue to the atoms of the actual
Transition State. Simple click on the atomname entries in the Z-matrix
editor and change them to the appropriate atoms. Click on the "apply
changes to current Z-mat" button for the changes to take
effect.
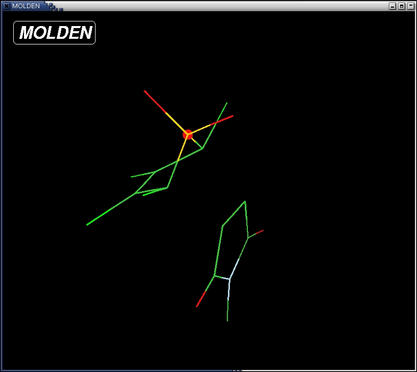
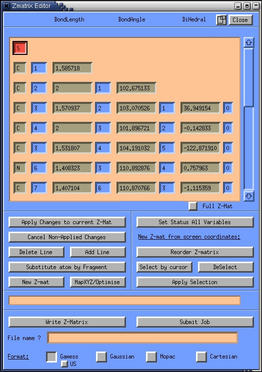
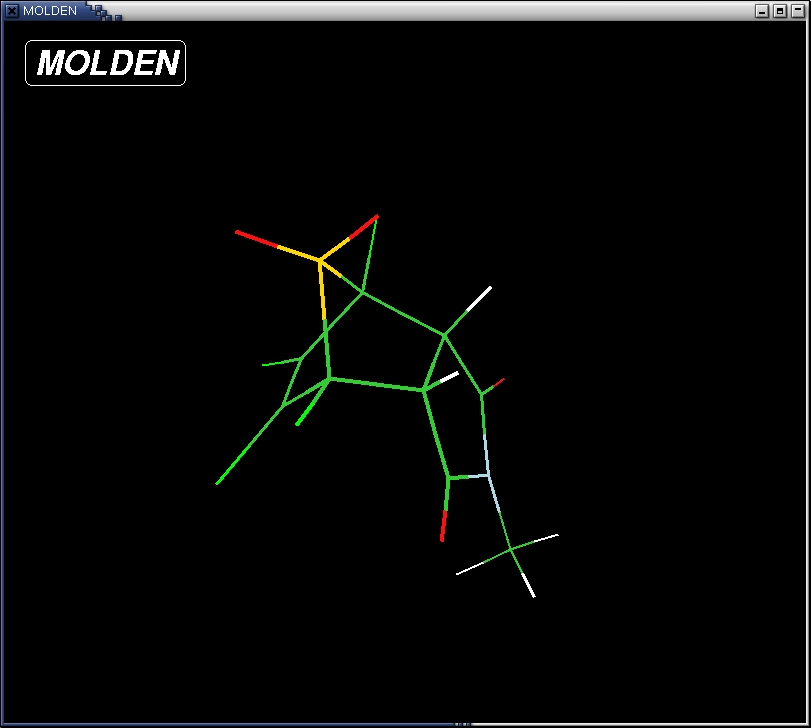
- saturate the complex with hydrogens. Use the "add line" button to
chose a hydrogen atoms and add them to the molecule by clicking on the
unsaturated atoms first followed by clicking two more atoms to define
an angle and a torsion respectively. The result should look like
this:
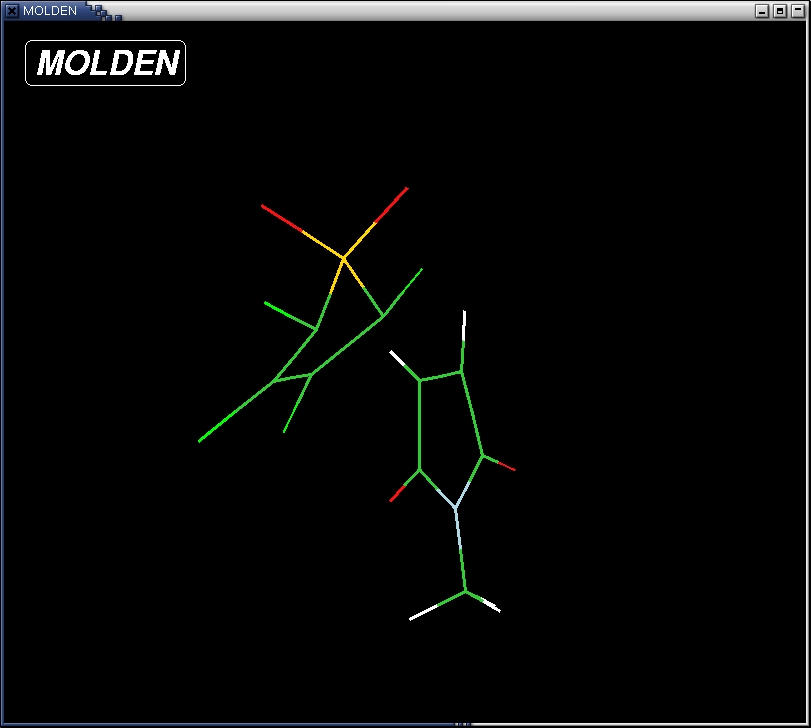
- save the Z-matrix into a gaussian format. Clicking the "Gaussian"
buttom at the bottom of the editor, type a name (e.g. TS.com)
in the "File name ?" window and click the "write Z-matrix"
button.
- quit molden by clicking the skull icon in the Molden Control window
- open analogue.pdb with MOLDEN
- The next step is to make an input file for Gaussian. We
use an ordinary text editor, such as emacs to complete the input file.
- open TS.com with emacs
localhost:~>rasmol 1C1E.pdb
- add the gaussian keywords for a transition state optimization (#P PM3 OPT=(TS,noeigentest) TEST) and compete the input file. We use a so-called checkpoint file (%chk=TS, first line) to store all optimized parameters. after the modification we save it as TS.com. An example of what the input file should look like is TS.com.
- open TS.com with emacs
- We run Gaussian to optimize the transition state.
-
localhost:~>g98 TS.com
-
copy the optimized cartesian coordinate section of the output file
(TS.log) and paste them into emacs. Modify the file such that it can
be read by Molden. The cartesian format for Molden is as follows:
line 1: number of atoms
line 2: blank
line 3: Atomsymbol x y z
line 4: Atomsymbol x y z
and so on. The last line needs to be blank!
save the file as TS.xyz. An example of what this file should look like is TS.xyz - open the cartesian coordinate file with the optimizaed transition
state structure with molden
localhost:~>molden TS.xyz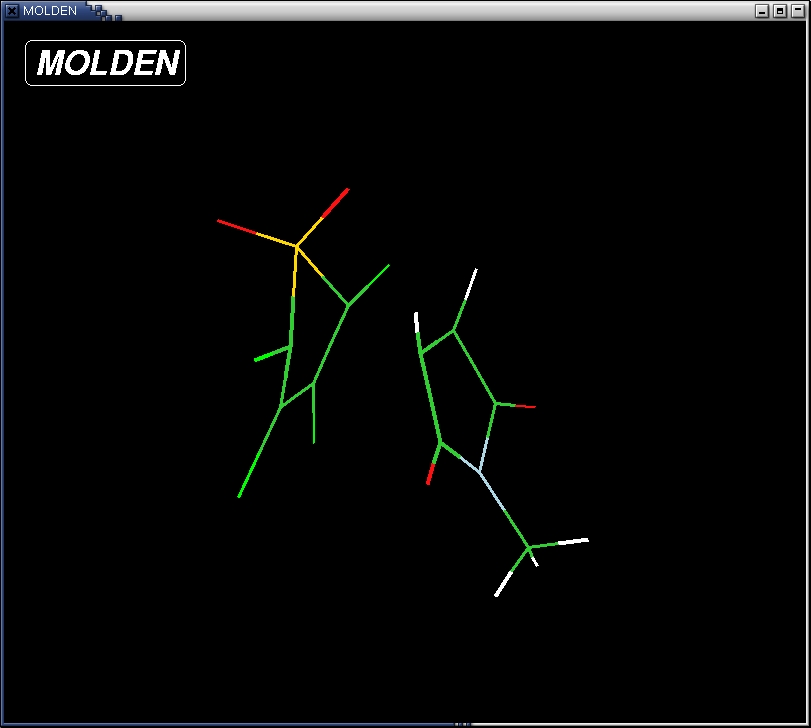
Note: the Nitrogen atom is not completely SP2 hybridized (planar) in this geometry, but rather a slightly SP3 hybridized (pyramidal). This is a known shortcoming of the semi-empirical PM3 hamiltonian. We ignore this artefact for now and continue with a slightly pyramidal N atom.
- We check if the structure obtained is a true transition state
- perform a freqency analysis on the optimized coordinates. The
frequencies, or normal modes, are the eigenvalues of the
HEssian. Remember that for a transition state, one and only one of
these eigenvalues needs to be negative. The inputfile for that looks
as follows:
%chk=TS
#P PM3 FREQ geom=check TEST
frequency calculation
0 1
The coordinates are read form the checkpointfile (TS.chk, geom=check). Save the file as TS_freq.com
-
localhost:~>g98 TS_freq.com
- check the output for imaginary freqencies. If there is one and only one, it means we found a transition state! From the visual inspection we learned it is the correct one, as the structure resembles what one would expect to be the transition state for the Diels-Alder cyclo-addition.
-
localhost:~>g98 TS.com